
微信公众号







2025版《中国药典》正式施行:对肽类原料与质量管理的三大影响
行业新闻动态

对于涉及肽类药物、功能性原料及特膳食品开发的企业而言,这次修订既是挑战,也是提升质量体系的机遇。本文将从三个角度解析新版《药典》对肽类原料和企业质量管理的核心影响。
新版《药典》延续了“与国际标准接轨”的导向,新增或修订了多项通用检测方法。在肽类及生物制品领域,尤以纯度、残留杂质及稳定性的检测要求最为关键。
纯度检测:强调采用高效液相色谱(HPLC)和质谱联用(LC-MS)等方法,对多肽片段的组成和比例进行定量检测,要求达到国际药典标准的一致性。
杂质控制:明确了肽类原料中的氨基酸衍生物、溶剂残留及微生物限度的检测范围,新增“单一杂质限值”指标。
稳定性验证:新版《药典》首次在说明性条文中提出,多肽类原料应进行温度-湿度双因素加速试验,以验证长期储存条件下的质量稳定性。
这意味着肽类产品从原料入厂、提纯、冻干到包装的每一个环节,都将面临更高标准的检测要求。企业需要同步更新实验室方法验证(Method Validation)及质控标准文件。
此次修订中,《药典》多处明确参考了**ICH(国际人用药品注册技术协调会)**及 USP(美国药典)的标准。例如,在通用技术要求中增加了“方法学可追溯性”的说明,要求实验室检验数据与国际指南标准保持一致性。
对企业而言,这带来三项直接影响:
研发环节需溯源:肽类原料的提取与纯化工艺需形成完整的工艺谱系(Process Lineage),数据可追溯。
质量体系需更新:ISO 9001、GMP 及 HACCP 体系文件要与新版《药典》检测要求同步修订。
供应商管理需强化:新增的“第三方检测机构备案制”意味着,企业应建立供应商与检测服务方的双重审核机制,确保原料检测结果具备法定效力。
这也是新版《药典》首次在制度层面对“数据一致性”(Data Integrity)提出硬性要求。未来的注册与备案审评中,数据完整性将成为审核重点。
随着新版《药典》的全面落地,肽类产品从研发到生产的监管强度将持续提高。企业应尽快完成以下三方面工作,以降低合规风险并抢占质量先机:
对照新版《药典》通用检测方法及相关条目,逐项梳理企业内部检测方法、标准品和检验记录,形成“差距清单”。
要求每一家原料供应商出具新版标准下的质检报告与方法验证文件,重点关注“批次一致性”和“检测方法来源”。
组织质量、注册、研发及市场部门进行联合培训,确保对新版《药典》核心要求有统一认知。所有宣传、资料、检测结果等对外发布内容,须经法务备案并注明数据来源,防止因“宣传用语不当”触发合规风险。
新版《药典》的实施,不仅标志着我国药品与功能原料标准体系迈向更高质量阶段,也对整个肽产业提出了更高要求。
对于肽类研究与生产企业而言,主动适配新版标准、强化数据溯源与质量验证体系,将成为企业长期稳健发展的关键路径。
监管趋严,但也让真正注重科研与品质的企业脱颖而出。
返回列表最新动态
 2024.07.24
2024.07.24
蒙肽制药2024团建
相聚辉腾锡勒草原,共享快乐时光

集团新闻
 2024.07.12
2024.07.12
推进骨肽乳酸菌口服液开发,
内蒙古“揭榜挂帅”项目再迎重要进展
集团新闻
 2024.07.05
2024.07.05
蒙肽制药:引领健康新篇章,荣获呼和浩特市
首家特殊膳食食品生产许可证
集团新闻
2024.09.27
蒙肽制药喜获
中国营养保健食品协会会员单位资格
集团新闻
 2025.04.11
2025.04.11
蒙肽制药
稳健走好每一步 专注国民补钙新布局
 2025.08.22
2025.08.22
蒙肽制药:揭榜挂帅项目“骨肽乳酸菌口服液开发产业化”圆满完成
 2025.09.29
2025.09.29
国家肉羊产业技术体系专家走进蒙肽制药——深化产学研合作,共话行业发展新未来
 2025.11.07
2025.11.07
药食同源产品联合研发签约仪式在呼和浩特举行
 2025.12.31
2025.12.31
携手奋进,共迎新年
 2017.04.17
2017.04.17
提醒|总局决定对新收到的35个药品注册申请进行临床数据核查
 2019.10.14
2019.10.14
一票制成医药行业发展趋势,
谁将受到较大影响?
行业新闻
 2019.10.14
2019.10.14
从仿制到创新,
中国医药行业转型突破口在哪?
行业新闻
 2019.10.14
2019.10.14
快年底了,
制药装备企业们都在忙些什么呢?
行业新闻
 2019.10.16
2019.10.16
数千亿的化学
仿制药注射剂市场将被“搅动”!
行业新闻
 2019.04.22
2019.04.22
4+7后跨国药企加速转型,
与国内药企合作成趋势!
行业新闻
 2017.01.22
2017.01.22
24家医药公司预计年报净利翻倍;国务院取消互联网药品交易资质审批
 2017.03.29
2017.03.29
倾情奉献 | 关注中药的朋友戳这里!近百位专家参与编写,约100册中药书籍年底问世!
 2017.01.09
2017.01.09
两票制国家文件正式定稿!2018年全国推开!
 2017.01.12
2017.01.12
听说,雾霾又要来了,制药企业这次还会被迫停产吗?
 2025.05.30
2025.05.30
行业动态盘点
2025年特殊膳食食品政策解读
 2025.07.18
2025.07.18
行业政策解读·2025年特殊膳食食品新规
 2025.08.08
2025.08.08
肽·分子营养学的新风口
 2025.07.11
2025.07.11
骨骼是行走的支柱:骨质疏松防治一网打尽
 2025.06.27
2025.06.27
多肽药物行业政策、监管与趋势解读
 2025.06.20
2025.06.20
骨骼健康相关的医药行业动态与政策解读
 2025.05.28
2025.05.28
骨胶原蛋白肽的前世今生——为什么它是骨骼健康的新风口?
 2025.08.19
2025.08.19
肽的全景指南:从分子到应用,带你看清“肽”这个赛道
 2025.08.19
2025.08.19
肽·分子营养学的新风口
 2025.08.29
2025.08.29
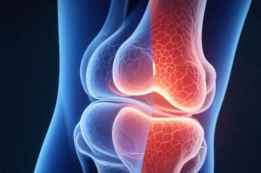
UC-II(未变性 II 型胶原)是什么?它如何影响关节的“炎症—软骨”循环

微信公众号

天猫旗舰店

抖音官方旗舰店
微信公众号

天猫旗舰店

抖音官方旗舰店

ICP备案:蒙ICP备案16005553号-2
蒙肽制药有限公司
网址:http://www.mengtaipharmacy.com
地址:内蒙古呼和浩特市和林格尔县盛乐经济园区燕京南街路北
电话:0471-3605001
邮箱:mengtaigroup@163.com